神经纤维瘤病诊疗指南(2025年版)
1.概述 神经纤维瘤病(neurofibromatosis,NF)是一组具有遗传异质性的神经皮肤综合征,是由基因缺陷所致的常染色体显性遗传病,可导致中枢或周围神经系统(包括脑、脊髓、皮肤和骨骼等)肿瘤。NF分为三种类型:占 NF 患者总数约 96% 的Ⅰ型神经纤维瘤病(neurofibromatosis type 1,NF1 )、约占 3% 的Ⅱ型神经纤维瘤病(neurofibromatosis type 2,NF2)和少于 1%的神经鞘瘤病(schwannomatosis,SWN)。
2.Ⅰ型神经纤维瘤病 2.1. 概述 NF1 是由 NF1(OMIM:613113)基因突变引起的常染色体显性遗传性肿瘤性疾病,外显率达 100%。患者多幼年起病,以多发咖啡牛奶斑(café-au-lait macules,CALMs )、神经纤维瘤为特征表现,可累及皮肤、神经、骨骼等多系统,肿瘤存在恶变风险。对于NF1 的管理、治疗仍存在挑战,多学科诊疗势在必行。
2.2. 病因和流行病学 NF1全球发病率约为 1/3000,家族性和散发病例各占半数。NF1是由肿瘤抑制基因NF1变异所致,新发突变率高,缺乏突变热点。NF1位于染色体 17q11.2 上,全长约350 kb,包含 57 个组成性外显子和 3 个可变剪接外显子,编码由 2 818 个氨基酸组成的胞浆蛋白——神经纤维蛋白,其广泛表达于神经元、黑素细胞、施万细胞等各种细胞。其带有中心 GTP 激活蛋白(GAP)结构域,可与 RAS-GTP 相互作用,显著提高固有的 GTP 活性,使其转化为 RAS-GDP,从而降低 RAS-GTP的水平,负向调节 RAF/MEK/ERK 和PI3K/AKT/mTOR 等信号通路,参与细胞分化、增殖及凋亡的调节,在肿瘤发生发展中起重要作用。在 NF1患者皮损内可发现自体的“二次打击”突变,可激活剩余的等位基因(即杂合性缺失),双等位基因 NF1 的激活已在神经纤维瘤的施万细胞和 CALMs 的黑素细胞中确认。NF1 基因变异则导致神经纤维蛋白表达下调甚至失活,RAS 通路异常活化,其下游分子RAF、MEK、ERK、PI3K、AKT、mTOR 表达上调,同时磷酸化水平上调,从而促进肿瘤的发生和发展。
2.3. 临床表现 NF1最常见的临床表现为CALMs 和神经纤维瘤。其他皮肤表现包括皱褶部雀斑、贫血痣、幼年黄色肉芽肿、蒙古斑周围“无色素晕”;神经系统表现包括认知障碍、学习困难、注意力缺陷多动障碍、癫痫、视神经胶质瘤(optic pathway glioma, OPG)、脑干胶质瘤、脊髓肿瘤、恶性周围神经鞘瘤(malignant peripheral nerve sheath tumor,MPNST)等;骨骼系统病变包括胫骨假关节、蝶骨翼发育不良、脊柱侧弯、身材矮小、巨头畸形、脊椎发育不良、腰椎翻转、漏斗胸等;其他表现如虹膜错构瘤(Lisch 结节)、青光眼、高血压、先天性心脏病、肺动脉高压、幼年型粒-单核细胞白血病(juvenile myelomonocytic leukemia,JMML)、嗜铬细胞瘤、乳腺癌、结直肠癌、胃肠道间质瘤、横纹肌肉瘤等的发生概率高于健康人群。该疾病存在较大的临床异质性,即使是同一家族的患者之间临床表现差异也可以很大。随着年龄的增长会出现完全外显的特征,半数患者在 1 岁时即可出现典型的临床表现,而其余患者到 8 岁时才可能基本满足,几乎所有患者在 20 余岁时均可达到诊断标准。
CALMs 几乎存在于所有 NF1 患者皮肤上,且通常是首发表现,约 99%的 NF1 患者于1 岁以内出现,常多发,散在分布于除掌跖外的周身皮肤。典型的CALMs 表现为浅褐色至深咖啡色圆形、卵圆形或不规则形斑片,边界清楚,直径数毫米到数厘米不等,大小随着儿童身体发育成比例增加。节段型NF1,又称镶嵌性NF1,是神经嵴来源细胞的 NF1 基因合子后突变体细胞和正常体细胞镶嵌所致,表现为局限于身体某一区域分布的 CALMs、雀斑和/或神经纤维瘤,病情通常比典型 NF1 温和。
神经纤维瘤是一类良性施万细胞肿瘤, 可分为皮肤神经纤维瘤(cutaneous neurofibromas,CNF)、皮下神经纤维瘤(subcutaneous neurofibromas,SNF)和丛状神经纤维瘤(plexiform neurofibromas,PNF)。CNF 最常见,起源于周围神经鞘,由含双等位基因突变的施万细胞增生而来。其瘤体由施万细胞、成纤维细胞、肥大细胞、内皮细胞和大量细胞外基质构成。一般出现自儿童期,表现为直径几毫米至几厘米的皮色橡胶状外生性软结节,数量和体积可随年龄增长而增加,数个至上千个不等,青春期和妊娠期是两个增长高峰,部分瘤体可引起瘙痒或疼痛。SNF 表现为皮下肿块,更坚硬,通常会感到疼痛,数量不等,也可多达成百上千,累及背根神经节的病变可能会压迫脊髓产生相应的临床症状。PNF 多在儿童时期发病,在NF1患者中的发病率达20%~50%,可沿神经轴索弥漫性生长,可累及头面部、眶内、四肢、胸腹盆腔、脊椎椎管内等,随年龄增长体积逐渐增大,如毗邻重要脏器常产生压迫症状,体积大的 PNF可能会导致皮肤、皮下组织和内脏的严重疼痛、毁容和运动障碍,瘤体内富含血管网,手术难以完整切除,甚至无法选择手术治疗。对于生长加速、疼痛、质地变硬等表现的神经纤维瘤应高度警惕 MPNST 可能,这是一种罕见的侵袭性肉瘤,可通过 PET 或活检以进行鉴别,对化疗和放疗相对耐受, 预后不佳,致死率较高,故也是 NF1 患者预期寿命较健康人群减少约 8~21 岁的主要原因。
目前已有部分基因型-表型相关性被发现。染色体微缺失通常表现出更严重的表型,多发性神经纤维瘤出现较早,且发生学习障碍、面部畸形、发育迟缓、心血管畸形,甚至MPNST 的风险更高。携带编码 Leu844、Cys845、Ala846、Leu847 和 Gly848 之一的结构性错义突变的个体,严重表型的发生率也较高,包括PNF、症状性脊髓神经纤维瘤、OPG、骨骼异常及恶性肿瘤。携带第 17 号外显子框内缺失突变(c.2970_2972delAATp.Met992del)的患者表型较轻微,主要表现为CALMs 和雀斑,无神经纤维瘤。影响 1809 位精氨酸密码子的 NF1 错义突变的患者也有较轻微的表型,表现为色素沉着特征,无神经纤维瘤、Noonan样特征。体细胞嵌合突变的患者除非合并 PNF,否则发生并发症的风险较小。全基因缺失的患儿各种症状出现的时间相较其他变异类型的患者都要早,病情更严重。
2.4. 辅助检查 2.4.1. 基因检测 可用于明确 NF1 诊断。应对临床诊断不明确、又需做出进一步诊疗或遗传咨询的疑似 NF1患者进行合适检测方案的基因检测以明确分子诊断、辅助制订疾病管理方案。首选检测方案为全外显子组测序,优先推荐先证者和/或父母行三人全外显子组测序。 根据NF1 的不同类型选择不同的送检样本,一般外周血即可,但节段型NF1 需对病变部位的组织进行检测。若检测结果仍为阴性,可考虑采用全基因组测序,并且每两年对原始数据进行重分析,以纳入与新发现的 NF1 相关基因或新突变等。基因检测也可辅助产前遗传学咨询。
2.4.2. 影像学 超声、X 线、CT、MRI 等影像学检查有助于骨骼、肿瘤等的诊断和检测。超声检查是筛查主要脏器病变及评估肿瘤最常用的检查,可用于原发瘤灶的评估及对治疗反应的监测。X 线平片检查可评估脊柱侧弯等骨骼异常情况。平扫及增强CT 用来评估原发肿瘤位置、范围及对周围组织侵犯情况,同时用于对治疗后的效果进行评估。MRI 或增强MRI 可确定原发瘤灶情况,以及其对周围邻近组织器官的侵犯情况,对 PNF 的诊断起到关键作用。PET-CT 检查用于全面评估瘤灶及全身伴发肿瘤情况,且有助于鉴别MPNST 伴转移与良性 PNF。
2.4.3. 组织病理学检查 对疑有恶变者,需要在原有的 PNF 内取多点活检来明确肿瘤是否发生了恶变,是 MPNST 诊断的金标准。
2.4.4. 眼科检查 直接检眼镜或裂隙灯检查可观察有无Lisch 结节,并与虹膜痣进行鉴别。还包括视力检查、对比视野检查、色觉检查,并评估瞳孔、眼睑、虹膜、眼底和眼外肌运动。
2.4.5. 智商测试及神经心理学评估 可以尽早发现认知缺陷,还有助于在学校为儿童提供学习支持。
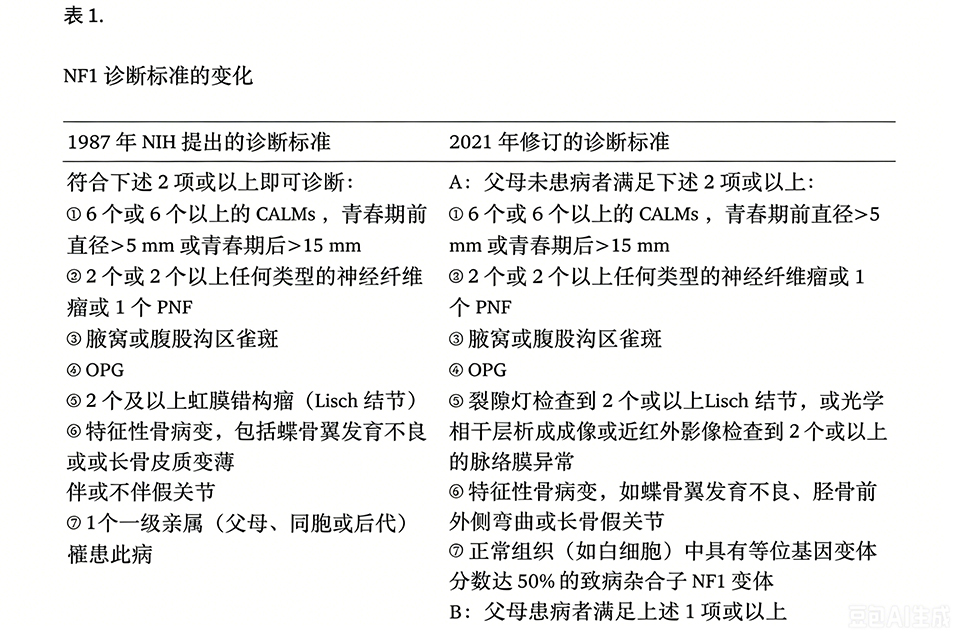
2.5. 诊断 既往 NF1 的诊断主要依据 1987 年美国国立卫生研究院(National Institutes of Health,NIH)提出的诊断标准。2021 年国际神经纤维瘤病诊断标准共识组对原诊断标准进行了修订(表1),主要增加了基因学诊断,将脉络膜异常加入眼科标准中,并提出同胞或子女患病不再纳入诊断标准的建议。修订后的诊断标准也被证实可以缩短 NF1 的诊断时间。
2.6. 鉴别诊断 多发CALMs 也可见于Legius 综合征、McCune-Albright 综合征、Noonan 综合征、结构性错配修复缺陷综合征等疾病。
① Legius 综合征:由 15 号染色体 SPRED1 双等位基因失活,同样导致 Ras-MAPK 通路上调,表现为典型的CALMs,可伴轻度雀斑、类似NF1 的认知障碍,但无神经纤维瘤或OPG。
② McCune-Albright 综合征:由 GNAS 基因体细胞突变引起,特别是 cAMP调节蛋白Gs α的突变,除了锯齿状 CALMs,还表现为多发性骨纤维性发育不良、内分泌亢进导致性早熟,且无神经纤维瘤。
③ Noonan 综合征:由Ras 信号通路中若干基因配体突变所致,半数患者存在PTPN11基因突变,表现为少量典型的 CALMs、肺动脉狭窄、身材矮小、面部特征明显、颈蹼。
④ 结构性错配修复缺陷综合征:是一种罕见的儿童肿瘤易感综合征,由 4 个错配修复基因(MLH1、MSH2、MSH6、PMS2)之一的双等位基因缺失突变引起,可表现为典型或非典型的 CALMs,本病具有高恶性风险如血液系统癌症、恶性胶质瘤。
色素沉着过度伴毛发增多的 PNF 也需与先天性黑素细胞痣鉴别。
NF1 仍需与NF2 进行鉴别,NF2 患者偶可见CALMs,无Lisch 结节,以双侧前庭神经鞘瘤、脑膜瘤、室管膜瘤等为特征。
基因检测有助于明确诊断。
2.7. 治疗 NF1 的治疗需要多学科联合治疗,涉及皮肤科、整形外科、肿瘤科、神经外科、放射科、眼科、骨科、神经内科、心内科、儿科、内分泌科、遗传学等。
2.7.1. CALMs 一般不必处理,对严重影响美观的皮损,可尝试激光对症治疗,如调 Q 激光、强脉冲光、点阵激光、皮秒、超皮秒。
2.7.2. CNF 治疗需结合患者意愿,对较大或对躯体功能造成影响的瘤体采取手术为主的治疗。术后可出现局部感染、瘢痕及复发等。瘤体数量较多、严重影响外观者也可采取CO2 激光消融、电干燥术、激光光凝术及射频消融术。药物治疗包括靶向 Ras-MEK 通路(司美替尼)、Ras-mTOR 通路、受体酪氨酸激酶等治疗尚待进一步研究,Ras-MEK 通路抑制剂外用制剂也在研发中。
2.7.3. SNF 及 PNF 对于有明显症状、恶变风险高、生长迅速及体积过大(直径>6 cm) 的神经纤维瘤应完善评估后,限期进行手术治疗,以预防恶变、改善外观、恢复功能等。根据肿瘤特征、部位和患者一般状况,选择全切除/近全切除(切除范围≥90%)、次全切除(50%≤切除范围<90%)或部分切除(切除范围<50%)。对于 PNF 的患者应进行长期、规律随访,并依据年龄、肿瘤部位、手术范围等对患者进行分级管理、肿瘤监测,指导手术的开展及术后复发风险的评估。PNF 的一线治疗以手术切除为主,对于有症状、无法手术完全切除的PNF 患者,也可应用MEK 抑制剂司美替尼靶向治疗,这是美国FDA 批准用于临床 PNF 的唯一治疗药物,70%用药者观察到部分缓解(肿瘤体积较基线时缩小≥20%,持续至少 4 周),80%患者疗效持续≥1 年,我国也已获批上市,常出现皮疹、甲沟炎等不良反应可耐受,对症处理即可。基因治疗、免疫疗法等目前仍处于研究阶段。
2.7.4. MPNST 应对患者进行全身评估,对于无远处转移征象的患者尽可能行早期手术治疗,远处转移的患者可选择放疗、化疗及靶向治疗。
2.7.5. 并发恶性肿瘤 应重视对肿瘤的早期识别和监测。不同恶性肿瘤的治疗原则和方案不尽相同。对于伴发 JMML 的患儿应尽快接受异基因造血干细胞移植,否则中位生存期短至10~12 个月。横纹肌肉瘤的治疗包括手术、化疗和局部放疗等多学科联合治疗,70%局限性患儿可被治愈。乳腺癌根据其病理类型、分期、分级、肿块大小及对激素敏感性等,选择手术、化疗、放疗、激素治疗、靶向治疗和免疫疗法。
2.7.6. OPG 治疗重点是视力保护,一线治疗方案为化疗,部分OPG 患者在未干预或一线化疗后可出现肿瘤消退。如伴有严重突眼影响外观、眶内段视神经肿瘤体积巨大,导致无光感,或继发暴露性角膜溃疡可考虑手术切除肿瘤。放疗虽有效,但由于可能出现严重不良反应,不作为首选推荐。靶向治疗仍在研究中。
2.7.7. 椎管内肿瘤 NF1 患者合并椎管内肿瘤的治疗原则同非 NF1 相关的椎管内肿瘤。如无相关症状且病情进展缓慢,可选择保守观察,严密随访。
2.7.8. 眼部病变 角膜神经纤维瘤以手术切除、角膜移植为主要治疗手段。角膜神经粗大患者可按需给予人工泪液治疗。NF1 合并青光眼患者视力预后差,首选药物降眼压治疗,若病情出现进展,可选择引流阀植入术、小梁切除术、房角切开术等手术治疗。脉络膜的NF1 改变无须治疗。
2.7.9. 骨骼异常 外科手术主要是对症治疗,缓解 NF1 患者由于骨骼异常带来的畸形和痛苦。若颅骨发育异常合并有邻近的占位性病变则需个体化考虑是否切除,以缓解占位性病变引起的颅骨异常的持续进展。对于脊柱侧弯,Cobb角 10°~25° 可采取运动疗法,25°~40° 可使用支具治疗和运动疗法,超过 40° 通常需采用融合或非融合技术,甚至截骨手术干预。颅骨异常引起脑膜膨出者建议早期外科干预。胸壁异常者,如心肺功能可以得到代偿, 往往无症状,仅因患者的美观需求而手术。NF1 患者若出现骨质疏松,建议补充钙片、维生素 D 及适度运动。如出现骨折可行复位固定,但再发骨折风险高,需注意预防。
2.7.10. 神经系统受累 NF1患者脑血管畸形、癫痫、头痛治疗原则同非 NF1 人群。
2.7.11. 心理、认知、生长发育异常 对于有言语功能障碍及运动功能障碍导致平衡和步态异常的儿童,应提供言语治疗、功能训练和理疗。当怀疑 NF1 患儿可能存在心理问题或精神障碍时,首先应该转诊到儿童心理或精神专科就诊或请会诊。然后根据具体情况制订相应的治疗方案,如心理治疗、药物治疗、物理治疗等。对于合并生长激素缺乏症的患者,在知情同意的基础上综合考虑获益、风险、患者治疗意愿、花费等,可试用注射用重组人 生长激素治疗,治疗中应密切监测相关不良反应,尤其是肿瘤发生风险。
2.8. 并发症监测 2.8.1. MPNST 建议NF1 患者每年进行 1 次皮肤科查体,在评估CALMs 和CNF 的同时, 还需评估有无 PNF,根据症状决策后续影像学检查或组织病理学检查。当原有的肿块大小或疼痛出现明显变化,或神经功能障碍迅速进展时,应警惕发生了恶性转化,尤其是肿瘤大小的变化最能预测肿瘤的恶性程度。超声检查可清楚显示肿块部位、大小、性质、血流情况、与周围组织毗邻关系、周围淋巴结是否肿大等。MRI 对于 MPNST 的诊断、临床分期、治疗及预后评估方面具有很高价值,为首选的影像学检查手段。筛查有无肺转移首选胸部CT 平扫,而骨扫描有助于判断有无骨转移。确诊MPNST 仍需要活检,推荐进行开放式且包括多个不同的肿瘤部位活检。建议每 3 个月随访 2 次,持续 3 年,之后 2 年内每 6个月随访 1 次,5 年以上每年随访 1 次。
2.8.2. 并发恶性肿瘤 若出现皮肤损害、发热、贫血、出血、肝脾大和肺部浸润等症状, 尤其是婴幼儿,需完善血常规、外周血涂片、骨髓细胞学检查结果、免疫分型、细胞遗传学特征、基因检测等,警惕 JMML。若头颈部、躯干四肢或泌尿生殖道等部位出现肿块, 或出现涕中带血伴鼻塞、外耳道脓性分泌物、吞咽困难、排便困难、血尿等,尤其是 2~5 岁或 15~19 岁患者,需完善超声、CT、MRI,肿块活检或手术切除,警惕横纹肌肉瘤。若在乳腺上摸到无痛性肿块,或同时伴有与月经周期无关的乳腺胀痛;或乳头溢液、酒窝征,或乳头皮肤瘙痒、糜烂、破溃、结痂、脱屑等,尤其是 25 岁以上患者,需完善乳腺超声、X 线钼靶、增强 MRI、活检,警惕乳腺癌。也建议 NF1 患者从 30 岁开始每年进行 1 次乳腺钼靶检查,并在 30~50 岁期间行乳腺 MRI 检查。
2.8.3. 并发颅内肿瘤 定期进行详细的神经系统体格检查。对儿童患者应监测视力变化直至8岁,出现可疑症状时应行头颅 MRI 检查,一旦诊断为 OPG 则需每年进行头颅 MRI 检查随访。
2.8.4. 椎管内肿瘤 早期无特异性临床表现,NF1 患者应每年常规行脊柱 MRI 检查。
2.8.5. 眼科病变 NF1 患者若出现视力下降、视野缺损、色觉改变时需要尽快请眼科医师进一步评估。确诊及疑诊 NF1 的 8 岁及以下儿童应至少每年做一次眼科检查,8~18 岁患儿可隔年一次,青春期后人群当出现可疑症状如视力下降应及时眼科就诊。年度随诊的主要项目包括:视力、视野、瞳孔反射、裂隙灯重点检查虹膜及眼底改变、色觉、眼球运动。 如果出现 NF1 相关眼部改变,需尽快安排光学相干断层扫描、视觉诱发电位、视神经及视交叉 MRI 检查。
2.8.6. 骨骼病变 NF1患者应每年进行全身骨骼系统评估,接受骨科体格检查,必要时行影像学检查,以了解躯干、四肢及颅骨发育异常的进展情况。
2.8.7. 神经系统受累 初诊及随访监测中的 NF1 患者均应接受详细神经系统体格检查。如疑诊脊旁神经根、神经干、神经丛等深部病变,应完善MRI、CT 识别病灶范围;如疑诊周围神经受累,予神经超声和电生理检查评估。
2.8.8. 心血管系统 对初诊及随诊中的 NF1 患者应进行详细的心脏查体。如可闻及心脏杂音,予超声心动图筛查先天性心脏病。NF1 患者至少在每年随诊时测量血压,并关注四肢血压、脉搏是否对称,听诊腹部有无血管杂音。对于合并高血压的儿童进行肾动脉和主动脉彩色多普勒超声筛查,对 30 岁以下成人进行肾动脉彩色多普勒超声筛查。对于血压难以控制者及有嗜铬细胞瘤相关表现者,完善血浆和尿液儿茶酚胺及其代谢产物水平等相关检测。NF1 患者也应预防性进行脑血管影像学筛查。
2.8.9. 心理、认知、生长发育相关 初诊及随访监测中的NF1 患者应注意发育进展。所有儿童或青少年期起病的 NF1 患者,均应在儿童期和青春期期间接受 1 次生长发育和心理评估。青春期前儿童应每年进行 1 次头围、身高、体重测量评估,从 5 岁起还需要每年评估第二性征性发育情况及线性生长情况以判定是否合并身材矮小、性早熟和青春期发育延迟。对于存在生长加速和/或性早熟患者,应行颅脑 MRI 检查评估有无下丘脑-垂体病变,尤其是 OPG。
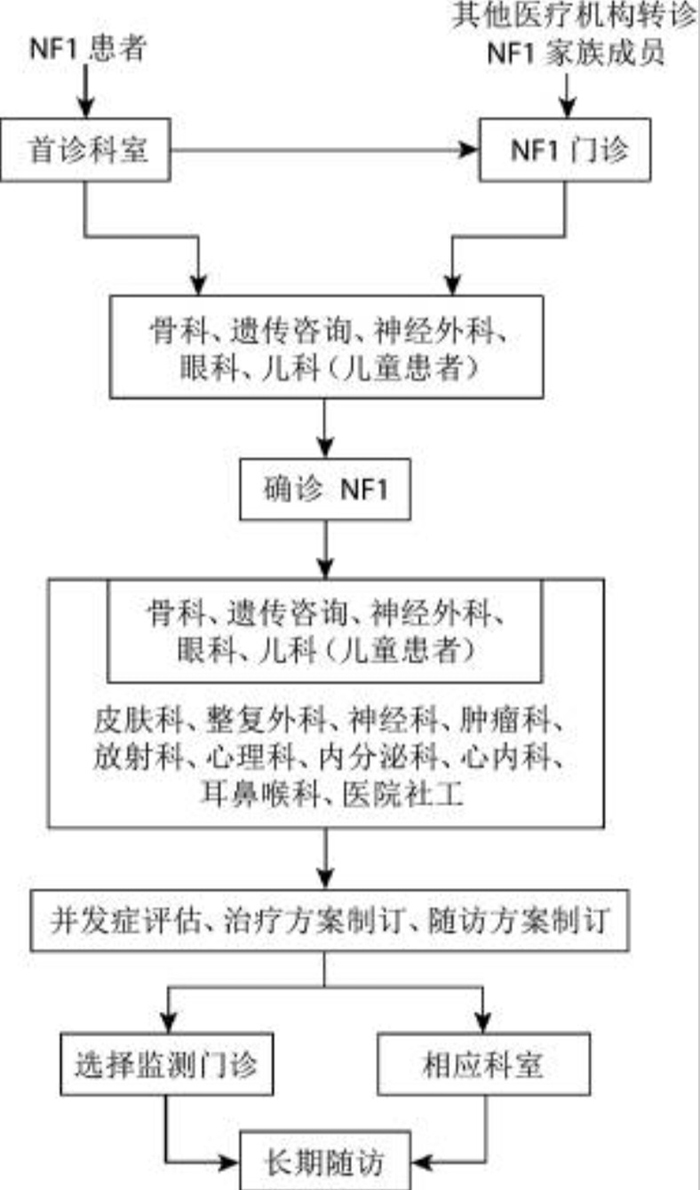
NF1多学科诊疗流程见图1。
- Ⅱ型神经纤维瘤病
3.1. 概述 NF2 约占NF 患者总数的 3%,是由位于染色体 22q12 上的 NF2(OMIM: 607379)抑癌基因杂合突变所致的常染色体显性单基因遗传病,发病率远低于 NF1,约为1/33000~1/25000。NF2 也具有广泛的表型异质性,病变涉及神经系统(神经鞘瘤、脑膜瘤、胶质瘤、室管膜瘤、星形细胞瘤、周围神经病变)、眼部(白内障、视网膜前膜病、视网膜错构瘤)和皮肤(皮肤肿瘤,最常见的是神经鞘瘤),其中双侧前庭神经鞘瘤为特征性表现,见于 95% NF2患者。
3.2. 病因和流行病学 NF2 由位于染色体 22q12上的 NF2 基因突变引起,超过半数患者是新发突变,其余为嵌合突变。抑癌基因 NF2 由 17 个外显子构成,编码分子量为 69 kD 的梅林蛋白,主要存在于神经系统组织中,负责细胞膜的稳定、参与多种细胞信号通路、调节多种细胞生长途径,磷酸化是其构象和肿瘤抑制活性调节的关键。与 Knudson 的肿瘤二次打击学说一致,即在遗传性肿瘤中,第 1 次突变来源于父母或生殖细胞,故是每个细胞都带有 1 个突变基因,在体细胞受到第 2 次突变,即该基因的两个等位基因失活时,异常或缺失梅林蛋白的细胞会在易受累的靶器官中形成肿瘤。
3.3. 临床表现 NF2 的早期症状通常出现在青年时期(20~30 岁),伴有前庭神经鞘瘤导致的听力损害,最初往往累及单侧,并可伴有耳鸣、头晕和听力失衡,后多发展为双侧,严重者导致耳聋、面神经功能减弱。在儿童患者中,约 30% 会出现听力损害,更常见的是视力障碍(白内障、错构瘤、颅内肿瘤)、皮肤肿瘤(斑块样病变最常见)、单神经病(面瘫、足下垂)、症状性脊髓肿瘤或非前庭颅内肿瘤。确诊后的存活期约为 15 年,平均死亡年龄为36~39 岁,10 年存活率为 67%。家系成员的表型和自然史相似,但家系间存在差异。
临床上将 NF2 主要分为两型:Gardner 型(轻型),症状较轻,发病晚,双侧前庭神经鞘瘤一般在成年时出现(平均年龄 22~27 岁),通常是唯一的特征。Wishart 型(重型),临床症状重,疾病进展快,除前庭神经鞘瘤外伴多发(且进展迅速)中枢神经系统肿瘤,其可能先于前庭神经鞘瘤出现,此型皮肤和眼睛受累也更明显。另外,还有一种类型被称为先天性 NF2,在出生前几天至几个月就能发现双侧前庭神经鞘瘤,这种类型可以长期稳定无症状,直到突然进展,可伴发非典型部位(如面部、手和脚)的肿块和其他中枢神经系统肿瘤(如脑膜瘤、室管膜瘤)。
3.4. 辅助检查 3.4.1. 影像学检查 增强 MRI 检查是 NF2 的首选检查,怀疑全身多部位病变时可行全身MRI 检查。MRI 可以发现直径小到 1~2 mm 的脑神经根和脊神经根肿瘤。前庭神经鞘瘤表现为实质性结节性肿块,边界清楚,明显强化。T1WI 呈等信号或低信号,T2WI 呈不均匀高信号,增强扫描病变实质部明显不均匀强化,病灶若有囊变、坏死则无强化,通常不存在钙化。另外亦可见来源于其他脑神经的神经鞘瘤,最常见于三叉神经。脑膜瘤常多发, 颅内任何部位均可发生,在 MRI 上表现为脊髓、脑或视神经周围脑膜上的明显均匀强化区域,可见脑膜尾征,脑膜瘤的生长速度比前庭神经鞘瘤更快。全脊柱 MRI 在高达 90% 的NF2 患者中检测到脊椎肿瘤,但只有 30%的患者临床上有脊椎肿瘤的症状。CT 对前庭神经鞘瘤的诊断起到补充作用,它能提供颅底,尤其是岩骨的解剖信息,有利于术前评估。
3.4.2. 眼科检查 用于识别特征性病变,如晶状体浑浊、视网膜错构瘤或视网膜前膜病。
3.4.3. 听力学检查 主要包括纯音测听、言语识别和脑干听觉诱发电位。在 10~72 岁的NF2 患者中,90%的患者存在纯音听阈异常。言语识别是功能性听力的衡量标准。脑干听觉诱发电位是一种更敏感的客观听觉功能指标,在有耳部症状的前庭神经鞘瘤患者中均是异常的,常表现为潜伏期延长。
3.4.4. 组织病理学检查 在诊断不明确的情况下,任何皮肤病变的活组织检查或其他病理学相关检查都可能是有帮助的。
3.4.5. 基因检测 虽然 NF2 基因检测不是诊断的必要条件,但是 NF2 基因突变种类可能会影响疾病的严重程度,基因型与表型存在一定的相关性。对基因型-表型相关性的研究发现, 一般来说,NF2 无义突变或移码突变的NF2 患者比错义突变或大片段缺失的NF2 患者病情更严重。在死亡率的相对风险中也存在基因型-表型效应,错义突变的患者比无义突变或移 码突变的患者死亡风险更低。
3.5. 诊断 既往NF2 的诊断一直沿用 1987 年制定的NIH 标准和 2005 年提出的Manchester 诊断标准。2019 年美国神经纤维瘤病会议修订了NF2 诊断标准(表2):满足A、B、C 诊断条件任意一项即可诊断。

3.6. 鉴别诊断 多发性脑膜瘤易与NF2 混淆,但不会出现神经鞘瘤的相关表现。部分儿童NF2 患者最初表现不典型,需与神经鞘瘤进行鉴别。一些罕见部位的神经纤维瘤易被误诊为 NF2,需术后病理明确。
3.7. 治疗 NF2 非常复杂,目前尚无有效治疗且可持续进展,因为肿瘤在手术切除后很可能复发,需要多学科协作,充分评估患者的整体状态、肿瘤负荷、治疗风险及获益后,制订个体化治疗方案。当患者出现脑干压迫、听力下降和/或面神经功能障碍时,仍需积极手术治疗,但也面临耳聋、面神经功能丧失、感染和头痛等手术风险。立体定向放射治疗可以作为手术的有效补充,但并不提倡治疗多发或巨大肿瘤。对于严重听力障碍的患者,采用耳蜗植入可能会获益。另外生物靶向治疗具体方案仍在研究中。
3.8. 并发症监测 ① 对于未经治疗的肿瘤患者,建议每年一次的 MRI 随访,为期 5 年,此后的随访间隔可以延长。
② 对于较大肿瘤,建议间隔缩小至6 个月以密切观察肿瘤大小。虽然5 年以后肿瘤再增长的概率下降,仍建议以较大的间隔进行影像学随访。
③ 对于保守治疗、放射治疗和不完全切除的患者每年进行 MRI 和听力测试随访,随访时间为 5 年。在肿瘤大小稳定的情况下,间隔时间可以增加一倍。
④ 对于肉眼肿瘤全切除患者,术后及术后 2、5、10 年 MRI 复查随访。
⑤ 对于已知存在 NF2 突变的个体,需每年进行 1 次听力评估(主要包括纯音测听、言语识别和脑干听觉诱发电位)、1 次眼科评估、1 次皮肤检查。10~12 岁开始监测 20 岁以下者全脑全脊髓 MRI,每 2 年重复 1 次,20 岁以后每 3~5 年重复 1 次。如果发现肿瘤, 检查频率应改为每年 1 次。
- 神经鞘瘤病 4.1. 概述 SWN 是最罕见的 NF 类型,属于常染色体不完全外显性遗传病,是起源于神经鞘膜施万细胞的良性肿瘤,又称施万细胞瘤,可发生于任何年龄,无明显性别差异,以全身多发性神经鞘瘤而不伴双侧前庭神经鞘瘤或皮内神经鞘瘤为特征,椎管内发病居多,可伴慢性疼痛、麻木、刺痛和虚弱。SWN 生长缓慢,边界清楚,常伴囊性变或退行性变,恶变可能较低。
4.2. 病因和流行病学 SWN 的发病率约为1/1700000~1/40000,诊断中位年龄约为 40 岁。其病因尚不明确, 在 40%~50%家族性和 10%散发性SWN 患者中发现 SMARCB1(OMIM: 601607)基因突变,且同时存在 NF2 的体系突变及失活(只存在于肿瘤局部,不会遗传给下一代),二者可能通过“四次打击-三个步骤”模式致病:首先是 SMARCB1 的种系突变(第 1 次打击), 之后是 22 号染色体的部分(包含第二个 SMARCB1 等位基因和一个 NF2 等位基因)丢失(第 2、3 次打击),然后另一个野生型 NF2 的等位基因突变(第 4 次打击)。散发病例中,NF2 突变的比例较 SMARCB1 更高。但部分SWN 患者仅存在 SMARCB1 的种系突变而无 NF2 的体系突变。在 SMARCB1 和 NF2 均无突变的病例中也发现了 LZTR 1(OMIM: 300574)的突变。部分家族性和大多数散发病例也无明确致病突变。家族性患者多携带非截短突变(错义或剪接为位点突变),散发患者更可能携带截短突变(移码或无义突变)。这些致病基因突变影响 Rac/PAK/JNK、Ras/Raf/MEK/ERK、PI3K/Akt/mTORC和 HDAC、Hippo/YAP、Wnt/β-catenin 等信号通路,促进肿瘤的形成。
4.3. 临床表现 SWN 患者最常在 20~30 岁出现症状,但正式诊断通常会推迟 10 年。患者通常出现疼痛、肿块或二者兼具。慢性疼痛通常与体位有关,与神经鞘瘤位置无关。SWN 常影响脊柱和周围神经,少数会累及脑神经(多为三叉神经)。由于肿瘤生长缓慢,起病隐匿,早期 一般无明显自觉症状或症状不典型,确诊时肿瘤体积往往已很大。5% SWN 患者会出现脑膜瘤,好发于大脑镰。SWN 患者没有学习障碍,极少数可发展为 MPNST。
4.4. 辅助检查 4.4.1. 影像学 CT、MRI 及超声均有助于明确诊断和估计瘤体大小、了解肿物与周围组织的位置关系,并能为术式选择及肿瘤切除范围的评估提供一定的依据。
4.4.2. 组织病理学检查 SWN 是梭形细胞构成的间充质肿瘤。在诊断不明确的情况下,任何皮肤病变的活组织检查或其他病理学相关检查都可能是有帮助的。
4.4.3. 基因检测 SWN 的诊断需要排除NF2 组成性突变,基因检测对诊断亦有辅助作用。
4.5. 诊断 SWN 的最新诊断标准见表3。

4.6. 鉴别诊断 由于 SWN 与 NF2 的临床表现存在重叠,故需与NF2 相鉴别,双侧前庭神经鞘瘤是二者鉴别的关键。但需注意嵌合型 NF2 也可能缺乏双侧前庭神经鞘瘤表现,仍需基因检测(血液及两个不同部位肿瘤的基因测序最为理想)等来确定诊断。
4.7. 治疗 SWN 患者的治疗经验有限,主要以症状为导向。针对疼痛,可使用加巴喷丁、普瑞巴林及短效阿片类药物和/或非甾体抗炎药,其他药物包括三环抗抑郁药如阿米替林、5-羟色胺-去甲肾上腺素再摄取抑制剂如度洛西汀、或抗癫痫药如卡马西平,可单独或联合应用。如疼痛控制不佳,可考虑手术切除疼痛性神经鞘瘤,这也是目前治疗有症状 SWN 的首选方法,可缓解局部疼痛或压迫症状,最大程度恢复神经功能。但术后疼痛复发较常见,且与肿瘤大小无关,也存在残留病灶导致肿瘤复发甚至恶变的可能。组蛋白去乙酰化酶抑制剂作为抗肿瘤药物,目前处于开发状态。
4.8. 并发症监测 SWN 的并发症一般是手术中或手术之后出现的。术中可出现出血、急性脑干梗死或机械损伤,重者可能发生术中死亡。术后初期易出现术后出血、颅内高压、脑干或小脑水肿。中晚期可能出现脑脊液漏、脑神经受损麻痹(面神经损伤最常见)、脑部症状(发音困难、共济失调)、锥体束受损。在术后晚期可出现颅内感染、脑脊液循环障碍、肿瘤复发等。
本文转载于中华人民共和国国家卫生健康委员会医政司网站
[1] Ghalavand MA, Asghari A, Farhadi M, et al. The genetic landscape and possible therapeutics of neurofibromatosis type 2[J]. Cancer Cell International, 2023, 23(1): 99.
[2] Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis[J]. International Journal of Molecular Sciences, 2021, 22(11): 5850.
[3] Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2[J]. The Lancet, 2009, 373(9679): 1974-1986.
[4] Blakeley JO, Plotkin SR. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis[J]. Neuro-Oncology, 2016, 18(5): 624-638.
[5] 谢超, 郭伟韬. 神经鞘瘤病的诊断及鉴别诊断[J]. 中国组织工程研究, 2015, 19(53): 8658-8659.
[6] 丁玉辉. 一个罕见椎管内神经鞘瘤病家系的临床与SMARCB1基因突变分析[D]. 天津: 天津医科大学, 2019.
[7] 王智超, 李青峰. Ⅰ型神经纤维瘤病临床诊疗专家共识(2021版)[J]. 中国修复重建外科杂志, 2021, 35(11): 1384-1395.
[8] 朱以诚. Ⅰ型神经纤维瘤病多学科诊治指南(2023版)[J]. 罕见病研究, 2023, 2(2): 210-230.
[9] 郭雅欣, 王鹤晓, 齐瑞群, 等. Ⅰ型神经纤维瘤病治疗新时代:司美替尼应用现状[J]. 中国皮肤性病学杂志, 2022, 36(12): 1344-1349.
[10] 国家卫生健康委员会. 儿童及青少年神经纤维瘤病诊疗规范(2021版)[EB/OL]. 2021.
[11] 2型神经纤维瘤病神经系统肿瘤多学科协作诊疗策略中国专家共识[J]. 中华神经外科杂志, 2021, 37(7): 663-668.